detk Quickstart¶
de_toolkit, or detk, is a python package and set of command line tools
that implements many common operations when working with counts matrices from
high throughput sequencing experiments. For example, the following diagram
illustrates various tools used in a simple RNA-Seq workflow downstream of
quantification:
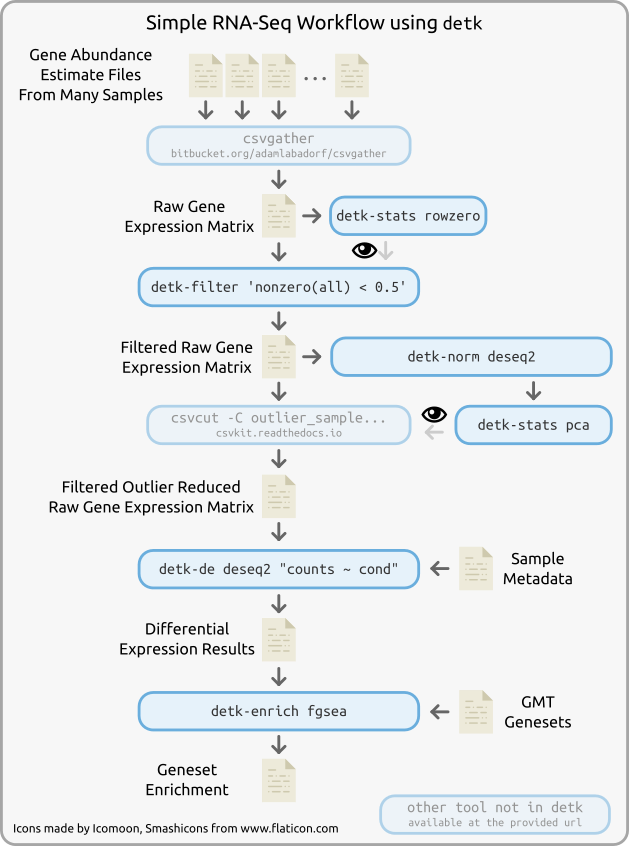
This workflow performs the following, all without any custom code:
- Takes the output from read counting (e.g. htseq-count) or expression estimation software (e.g. salmon or kallisto) and combines them into a concatenated counts matrix using the csvgather tool
- Calculates statistics on the zero-ness of genes that is helpful in making decisions for filtering features using detk-stats rowzero
- Filter out rows from the raw expression matrix that have half or more zero counts detk-filter
- Normalizes the filtered counts matrix file using the DESeq2 normalization procedure detk-norm deseq2
- Computes and visualizes principal components on the normalized counts matrix to identify outlier samples detk-stats pca
- Uses the
csvcuttool from the csvkit software package to remove a hypothetical outlier sample from the raw matrix - Conducts differential expression using the DESeq2 method by combining the raw counts matrix with a sample metadata file detk-de deseq2
- Computes pre-ranked GSEA analysis on the differential expression statistics using the fgsea package
These tools were designed by and for analysts who implement analyses like this one regularly on the command line. The above workflow could be easily implemented with common workflow management software like snakemake like so:
from glob import glob
rule all:
'detk_report/detk_report.html',
'msigdb_c2cp_gsea_results.csv'
rule gather_counts:
input: glob('sample_*__salmon_counts/quant.sf')
output: 'raw_counts.csv'
shell:
'''
csvgather -j 0 -f NumReads -f "s:NumReads:{{dir}}:" \
-f "s:__salmon_counts::" -o {output} \
{input}
'''
rule raw_rowzero:
input: 'raw_counts.csv'
output: 'raw_counts_rowzero_stats.csv'
shell:
'detk-stats rowzero -o {output} {input}'
rule filter_raw:
input: 'raw_counts.csv'
output: 'raw_counts_filtered.csv'
shell:
'detk-filter "nonzero(all) < 0.5" -o {output} {input}'
rule deseq2_norm:
input: 'raw_counts_filtered.csv'
output: 'norm_counts_filtered.csv'
shell:
'detk-norm deseq2 -o {output} {input}'
rule pca:
input: 'norm_counts_filtered.csv'
output: 'norm_counts_filtered_pca.csv'
shell:
'detk-stats pca -o {output} {input}'
rule generate_detk_report:
input:
rules.raw_rowzero.output,
rules.pca.output
output: 'detk_report/detk_report.html'
shell:
'detk-report generate --dev'
rule remove_outlier:
input: 'raw_counts_filtered.csv'
output: 'raw_counts_filtered_nooutlier.csv'
shell:
'csvcut -C outlier_sample_name {input} > {output}'
rule de:
input:
counts='raw_counts_filtered_nooutlier.csv',
covs='sample_info.csv'
output: 'deseq2_results.csv'
shell:
'detk-de deseq2 -o {output} "counts ~ cond" {input.counts} {input.covs}'
rule gsea:
input:
de='deseq2_results.csv',
gmt='msigdb_c2cp.gmt'
output: 'msigdb_c2cp_gsea_results.csv'
shell:
'detk-enrich fgsea -o {output} -i gene -c cond__log2FoldChange {input.gmt} {input.de}'